1. 临床期间,药品对患者安全性考量因素更重,分析方法需要进行适当的验证才能进一步保障患者的安全性。GMP附录《临床试验用药品(试行)》,在放行中就有对分析方法验证状态的审核,所以肯定要是做的。
2. GMP附录《临床试验用药品(试行)》,也说明了临床试验期间,通常生产工艺还不够成熟,对药品特性还需要进一步了解,所以还不具备充分验证的条件。所以一般只进行部分验证。
3. 临床阶段患者安全性考量更重,所以更多考虑安全性控制项目的验证项目,如专属性、灵敏度等。等工艺成熟了,具备充分验证条件了再完整验证(这时候通常临床已经做了。)具体可能参考《2023版药品GMP实施指南》(质量管理体系)。
GMP附录《临床试验用药品(试行)》:

GMP附录《临床试验用药品(试行)》:

《2023版药品GMP实施指南》(质量管理体系):

1. 参照2020年《新药I期临床试验申请技术指南》相关要求研究并制定临床试验样品质量标准,以表格形式提供相关检测项目和可接受限度要求。注意提供关键项目(例如有关物质、残留溶剂、金属催化剂残留、致突变杂质、溶出度/释放度、含量等)的具体分析方法,以及必要的方法学验证信息(例如至少包括专属性、灵敏度等)。
提供关键研究批次(包括用于安全性研究、稳定性研究、临床研究(如已制备)等批次)的检验报告。
2. 2023年4月26日,药品审评中心培训《新药临床试验申请(IND申请)前药学沟通交流技术要求及案例分析》:
共性问题:仅简单提供质量标准,缺少提供部分关键项目的分析方法、未提供关键项目分析方法必要的验证信息。
一般要求:参照《新药1期临床试验申请技术指南》相关要求提供信息。注意提供关键项目(例如:有关物质、 残留溶剂、 金属催化剂残留、 致突变杂质、溶出度/释放度、 含炬等)的具体分析方法, 以及必要的方法学验证信息(例如至少包括专展性、 灵敏度等) 。(IND阶段不要求迸行全面的方法学验证, 但基本保证能看得见)
提供关键研究批次(包括用于安全性研究、 稳定性研究、 临床研究(如己制备)等批次)的检验报告。 (关注质量不熊桥接时的潜在安全性风险)。
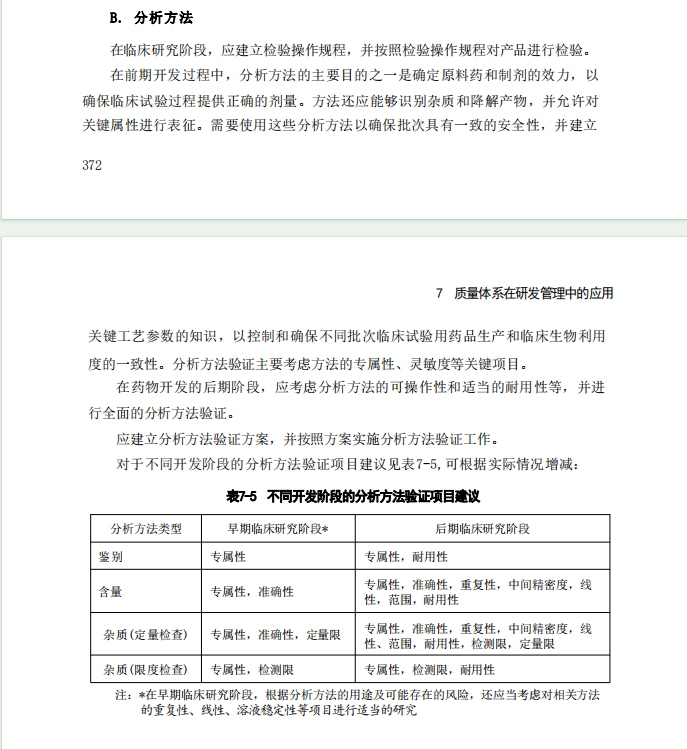
临床申报要分不同阶段去考虑。
- 比如做申请 I 期临床,验证关键项即可。
依据可参考《新药I期临床试验申请技术指南》
1.1.5质量控制
应提供初步的质量标准,说明检查项目、可接受的限度、分析方法,提供代表性图谱。在药品开发初期,不需要提交全面完整的分析方法验证资料,但至少应提供方法的专属性、灵敏度等关键验证信息。
依据可参考《药品生产质量管理规范(2010年版)》第二百二十三条
(一)企业应当确保药品按照注册批准的方法进行全项检验;
(二)符合下列情形之一的,应当对检验方法进行验证:1.采用新的检验方法;2.检验方法需变更的;3.采用《中华人民共和国药典》及其他法定标准未收载的检验方法;4.法规规定的其他需要验证的检验方法。
(三)对不需要进行验证的检验方法,企业应当对检验方法进行确认,以确保检验数据准确、可靠。
这{{threadTextType}}正{{isAdminText}}
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: