【建议】
1. 如果生成了研究报告,递交DSUR或年报的时候,需要递交报告和SEND数据包。只要是这个新的报告也是2017年12月之后做的,是属于“单次给药毒性、重复给药毒性、安全药理学(呼吸、心血管)和致癌毒”研究里面的,CDER都要求有SEND datasett跟着这个报告,否则拒收(触发rejection条件)。
2. 如果届时没有生成研究报告,那就是会在文件说明今年做了xx长毒实验,summary一下,后面报告生成了再用amend形式递交上去,等amend递交的时候,还是需要制作SEND。
【依据】
1. 21 CFR Sec.314.81(2) annual report
(v) Nonclinical laboratory studies. Copies of unpublished reports and summaries of published reports of new toxicological findings in animal studies and in vitro studies (e.g., mutagenicity) conducted by, or otherwise obtained by, the applicant concerning the ingredients in the drug product. The applicant shall submit a copy of a published report if requested by FDA.
5. 非临床研究。申请人从开展的与药品成分相关的动物实验和体外研究(例如致突变实验)中,或申请人收集到的新毒理学发现的未发表的研究报告以及发表的研究报告摘要的副本。如FDA要求,申请人需递交完整的已发表的研究报告的副本。
2. FDA. Technical Rejection Criteria for Study Data
3. FDA. Guidance for Industry Providing Submissions in Electronic Format -- Standardized Study Data
非临床数据交换标准(Standard for Exchange of Nonclinical Data,SEND),是临床数据交换标准联盟(CDISC)基于临床研究数据制表模组(SDTM)的拓展开发,针对非临床试验数据提供一致性的架构,包括数据格式,变量的标准名称等,允许个别毒理试验数据以标准化和电子化方式呈现。电子数据格式标准化有助于药品监管机构能够有效接受、处理、审查和归档申办方提交的资料,提高审查效率。
SEND文件是eCTD子文件夹中必须包含的内容,主要分为SEND 3.0、SEND 3.1和SEND DART三个版本。FDA针对各版本强制实施的日期及内容如下表所示。
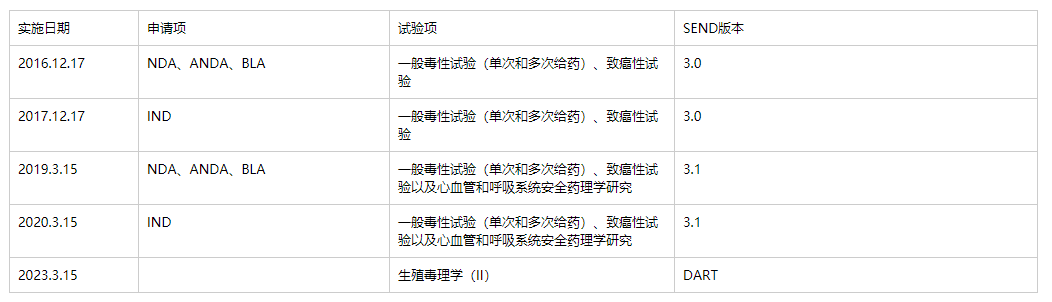
Deadlines: Sponsors whose studies started after December 17, 2016, must use the data standards listed in the FDA Data Standards Catalog for NDAs, BLAs and ANDAs. For Commercial INDs, the requirement applies to studies started after December 17, 2017.
对于NDA/BLA/ANDA的申请,如涉及2016年12月17日之后开展的如上表列出的非临床研究;对于IND申请,如涉及2017年12月17日之后开展的如上表列出非临床研究,申报资料必须采用SEND格式。
2021年9月15日以后申报IND,要求同时递交安全药理(心血管和呼吸系统)和关键毒理的SEND数据包,否则无法受理。

这{{threadTextType}}正{{isAdminText}}
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: