对于ANDA,已得到正式回复,也分享给大家。如下:
药典标准是法定最低标准,产品注册申报时是否必须制定有关物质检查项还是要根据具体产品具体讨论;在药典拟定的质量指标不足以对杂质尤其是降解杂质控制的情况下,是不能被接受的。
法规/指南依据总结如下:
1)根据ICH Q3B(R2)Impurities in New Drug Products(FDA已作为工业指南发布):
The
applicant should summarize the degradation products observed during manufacture
and/or stability studies of a new drug product. This summary should be based on
sound scientific appraisal of potential degradation pathways in the new drug
product and impurities arising from the interaction with excipients and/or the
immediate container closure system. In addition, the applicant should summarize
any laboratory studies conducted to detect degradation products in the new drug
product. This summary also should include test results of batches manufactured
during the development process and batches representative of the proposed
commercial process. A rationale should be provided for exclusion of those
impurities that are not degradation products (e.g., process impurities from the
drug substance and impurities arising from excipients) ….
要求:申请人总结在新药产品的生产和/或稳定性研究过程中观察到的降解产物,且应基于对新药产品中潜在降解途径和与辅料和/或直接容器密封系统相互作用产生的杂质的合理科学评估。此外,申请人应总结为检测新药产品中的降解产物而进行的任何实验室研究。还应包括在开发过程中生产的批次的测试结果和代表拟议商业过程的批次。应提供排除那些不是降解产物的杂质的理由(例如,来自原料药的工艺杂质和来自赋形剂的杂质)……
ICHQ6A Specifications: Test Procedures and Acceptance Criteria for New Drug Substancesand New Drug Products: Chemical Substances(FDA已作为工业指南发布),在章节3.2.2.
New Drug Products中提及到:
Organic
impurities arising from degradation of the new drug substance and impurities
that arise during themanufacturing process for the drug product should be
monitored in the new drug product.
明确了:新药降解产生的有机杂质和制剂生产过程中产生的杂质应在新药中进行监测。
3)根据FDA工业指南,ANDAs:Impurities in Drug Products提及:
We
recommend that you include in your submission a rationale for the inclusion or
exclusion of degradation products in the drug product specification. It is
important that the rationale include a discussion of the degradation profiles
observed in stability studies and any other batch(es) manufactured in support
of the application.
FDA“建议”:申请人在提交的文件中要包含在药品质量标准中包含或排除降解产物的理由。重要的是,基本原理包括对稳定性研究中观察到的降解曲线的讨论以及为支持应用而制造的任何其他批次。
根据上述指南要求,稳定性研究数据应该是有限的,建议初始注册申报时对仿制药产品中的每个特定杂质、任一非特定杂质和总杂质进行控制,以进行放行测试和稳定性研究。
在获得足够数据证明相关物质不是降解产物后,可以提交批准后的事先批准补充
(PAS)进行变更,以取消产品质量标准中的有关物质检查项。
对于FDA注册申报来说,USP是最低门槛,如果USP都不能符合的话对于能FDA注册申报的概率就很小,在满足了USP的基础上,也是需要满足其他的法律法规、行业指南的要求,例如ICH Q3,Q6,M7等;
根据MAPP 5017.2 Establishing Impurity Acceptance Criteria As Part of Specifications for NDAs, ANDAs, and BLAs Based on Clinical Relevance,要求原料药有关物质需要包括的检测项目如下:
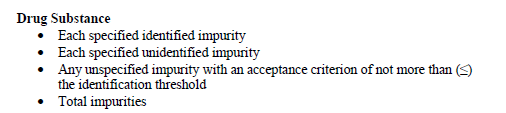
再结合QbR对于原料药质量标准的要求:
Q:What if there are no impurities' tests found in the USP monograph for a USP drug substance? What should the ANDA sponsor do?
A:Please work with your supplier (DMF Holder) to ensure that potential synthetic process impurities (e.g. isomers (if any), side reaction products), degradation impurities, metal catalysts, and residual solvents are adequately captured by your impurities test method. There may be information available in published literature as well, regarding potential impurities.
综上:不要投机取巧,在研发初级阶段就应该做好相关杂质的研究,以及相关的方法验证和稳定性考察;如果实在是不想在初次申报就报那么多杂质,想按照USP药典来赌一赌的话也是可以的,不过相关的研究需要做充分了。退一步讲,这种做法制剂客户应该都不会同意的
这{{threadTextType}}正{{isAdminText}}
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: