建议:
1、可参考已上市化学药品药学变更研究技术指导原则(试行)中三、变更原料药生产工艺综述提及:
在本指导原则中,当对比研究结果符合以下条件时,则可认为杂质谱一致:
①新增杂质未高于《化学药物杂质研究的技术指导原则》 及 ICH Q3A 等规定的鉴定限度;
②已有杂质(包含立体异构体)及杂质总量均在质量标准规定的限度内,如标准中无规定,应在原工艺生产的多批产品测定范围内;
③新使用的溶剂残留量符合《化学药物有机溶剂残留量研究的技术指导原则》及 ICH Q3C 等的有关规定;
④新的无机杂质符合《化学药物杂质研究的技术指导原则》及 ICH Q3D 等的有关要求。⑤应参考 ICH M7 对致突变杂质进行考察,必要时进行控制。
2、可参考80号文中注册分类4、5.2类申报资料要求(试行)32P5提及的列表提供:
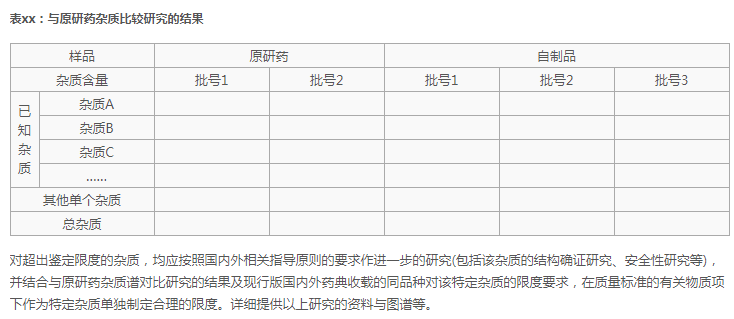
依据:
思考:
杂质的研究本身就可深可浅,因此杂质谱的对比至少应包括质量标准控制的相关杂质,往深了,可以对比所有检出的杂质,清晰可见杂质检出情况。
CDE变更指导原则试行版总结了有机杂质,溶残,基度以及金属元素等同的要求,对比了和其他市场的要求,已经比较全面,可以供参考,具体如下。
国内要求:
根据已上市化学药品药学变更研究技术(试行)2021.02版本,当对比研究结果符合以下条件时,则可认为杂质谱一致:
①新增杂质未高于《化学药物杂质研究的技术指导原则》 及 ICH Q3A 等规定的鉴定限度;②已有杂质(包含立体异构体)及杂质总量均在质量标准规定的限度内,如标准中无规定,应在原工艺生产的多批产品测定范围内;③新使用的溶剂残留量符合《化学药物有机溶剂残留量研究的技术指导原则》及 ICH Q3C 等的有关规定;④新的无机杂质符合《化学药物杂质研究的技术指导原则》及 ICH Q3D 等的有关要求。⑤应参考 ICH M7 对致突变杂质进行考察,必要时进行控制。
美国市场参考2018.09 Postapproval Changes to Drug Substances Guidance for Industry
Equivalence of Impurity Profiles
The impact of manufacturing modifications on the impurity profile (including impurities addressed by ICH M7) is evaluated by determining levels of existing and new impurities. It is important to determine the stage in the manufacturing process at which impurities should be evaluated and to establish the adequacy of the analytical procedures used for this purpose. Levels of residual solvents and inorganic substances should also be considered during evaluation of the impurity profile.
If the impurity profile of an isolated material (i.e., isolated intermediate, unfinished drug substance, or drug substance) following the change is equivalent to that of pre-change material, the drug substance's impurity profile will be considered unaffected by the modification. If the manufacturing modification occurs at an upstream step before the final intermediate is produced, and equivalence cannot be demonstrated for the intermediate isolated immediately following the change, the impurity search should be extended to the next downstream intermediate. The impurity search should also be expanded to include appropriate downstream impurities that may be formed during the manufacturing process. The evaluation process should be repeated on downstream intermediates up to and including the drug substance. Equivalence should not be established by mixing pre- and post-modification materials or materials from different batches during manufacturing operations.
http://lib.shilinx.com/wiki/index.php?title=FDA_Postapproval_Changes_to_Drug_Substances_Draft_201809 (识林链接)
ICH Q11 Q&A
这{{threadTextType}}正{{isAdminText}}
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: