【建议】
多剂量的口服混悬液,在药液浓度相同的情况下,50mL和100mL只是包装规格的不同。可生产3批药液,并将药液灌装成50mL和100mL共6批供试品,进行稳定性研究,不必生产6批药液。
【依据】
(1)根据FDA ANDAs: Stability Testing of Drug Substances and Products, Questions and Answers Guidance for Industry(ANDA:原料药和制剂稳定性试验 问题与解答)——
Section C Drug Product Manufacturing and Packaging (C. 制剂生产和包装)
Q19(i): In cases where an intermediate bulk material is identical between the various strengths (dose proportional blends, bulk solutions, etc.), is it sufficient to perform stability on one lot of each strength, when each strength is produced from a separate intermediate bulk?
Q19 (i): 在各个规格的(剂量比例混合物、原液等)之间的中间体散装物料相同的情况下,当每个规格由独立的中间体物料制得,是否仅对每个规格取一批开展稳定性研究就足够了?
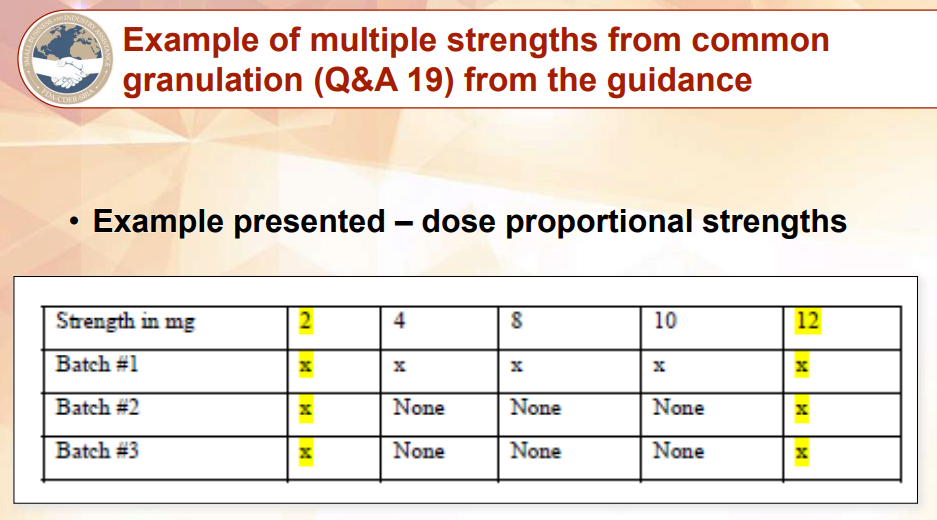
A19(i): No. For ANDAs that contain multiple strengths (that are dose proportional), three separate intermediate bulk granulations (or blends) should be manufactured. One batch of bulk granulation (or blend) should be used to manufacture all the strengths proposed. The other two bulk granulations (or blends) can be used to manufacture only the lowest and the highest strengths, in addition to the strength used in BE studies (i.e., the strength(s) tested in the BE studies should have three batches). Stability testing should still use all three batches of drug product.
A19(i): 不可以。对于包括多个规格(按剂量成比例)的ANDA,应生产三个单独的中间体散装颗粒(或混粉)。一批颗粒(或混粉)应被用于生产所有拟议规格的产品。另外两批散装颗粒(或混粉)可仅被用于生产最低和最高规格的产品,生物等效性研究使用的规格除外(即, BE 研究中检测的规格应该有三个批次)。稳定性试验仍应使用所有三批药品。
此外,还需要参考C部分的
Q1: Can the split bulk solution filled into different fill volumes be considered discrete batches?
Q1: 将散装溶液分装灌入到不同灌装体积可以被认为是不同的批次吗?
A1: To be consistent with ICH Q1A(R2), we recommend that discrete finished product batches be produced that represent different batches of bulk solution. Split filling one batch of bulk solution into different fill volume sizes would not constitute discrete batches.
A1: 为与ICH Q1A(R2) 保持一致,我们建议生产代表不同批次散装溶液的非连续成品制剂批次。将一批散装溶液分装至不同灌装体积不构成非连续批次。
(2)根据CDER法规培训ANDA Stability Guidance(s)(https://www.fda.gov/media/91781/download)提到:参考问答指南C部分Q19,对于中间物料规格的稳定性研究,可采用括号设计法。

下图即为(1)中A19(i)的举例:
(3)根据ICH Q1D:
2.3括号法 是一种稳定性试验的设计方案,它仅对某些设计因子(如规格、包装容器大小或装量)处在极端状态的样品,与完整设计方案一样,在所有时间点进行试验。这种方案假设:任何中间状态样品的稳定性可以用被试验的极端状态样品的稳定性所代表。
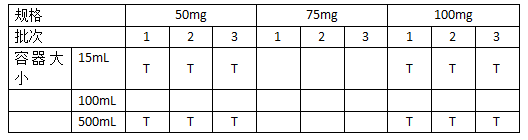
2.3.1.1 规格:括号法可用于处方相同或相近的多个规格样品稳定性研究中。例子包括但不限于(1)由相同粉末混合物、不同填充量制成的不同规格的胶囊;(2)由不等量的同种颗粒压制成的不同规格的片剂;和(3)处方仅在某些微量辅料(如着色剂、矫味剂)上有差别的不同规格的口服溶液剂。经过验证,可将括号法应用于处方中原辅料比例不同的多个规格的研究中。这样的验证可以包括在临床试验或产品研发中所用批次的产品不同规格间稳定性情况的比较结果。如果各规格之间使用了不同的辅料,就不能应用括号法。
2.3.1.2 容器大小和或装量:可将括号法应用于容器大小或者装量不同而其他保持不变的同种包装容器系列的研究。但是,如果在容器大小和装量均发生变化的情况下。考虑使用括号法,就不能假设最大和最小的容器代表了所有包装形态的极端状态。应通过比较包装容器系统中可能影响产品稳定性的各种特性来仔细选择包装形态的极端状态。这些特性包括容器壁厚度、闭塞物的几何形状,表面积与体积之比,上部空间与总体积之比,每个剂量单位或单位装量体积的水蒸气透过率或氧气透过率等。经过验证,括号法可以用于同种容器、不同闭塞物的研究。验证内容可以包括该系列容器密闭系统的相对通透率的讨论。
括号法举例:
【思考】关于这个问题的解答还有个引出来的问题想探讨,没弄太清,即“1批药液分装成2个包装,申报时是按照1个规格报还是2个规格?”
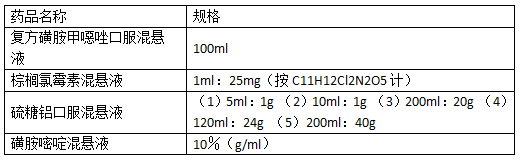
中国药典2020版二部凡例第二十条表述,“制剂的规格,系指每一支、片或其他每一个单位制剂中含有主药的重量(或效价)或含量(%)或装量。注射液项下,如为“1ml:10mg”,系指1ml中含有主药10mg;对于列有处方或标有浓度的制剂,也可同时规定装量规格。”这种表述我还是不太理解。通过查阅药典记载的混悬液药品进行比较,如下表,各种规格描述都有。
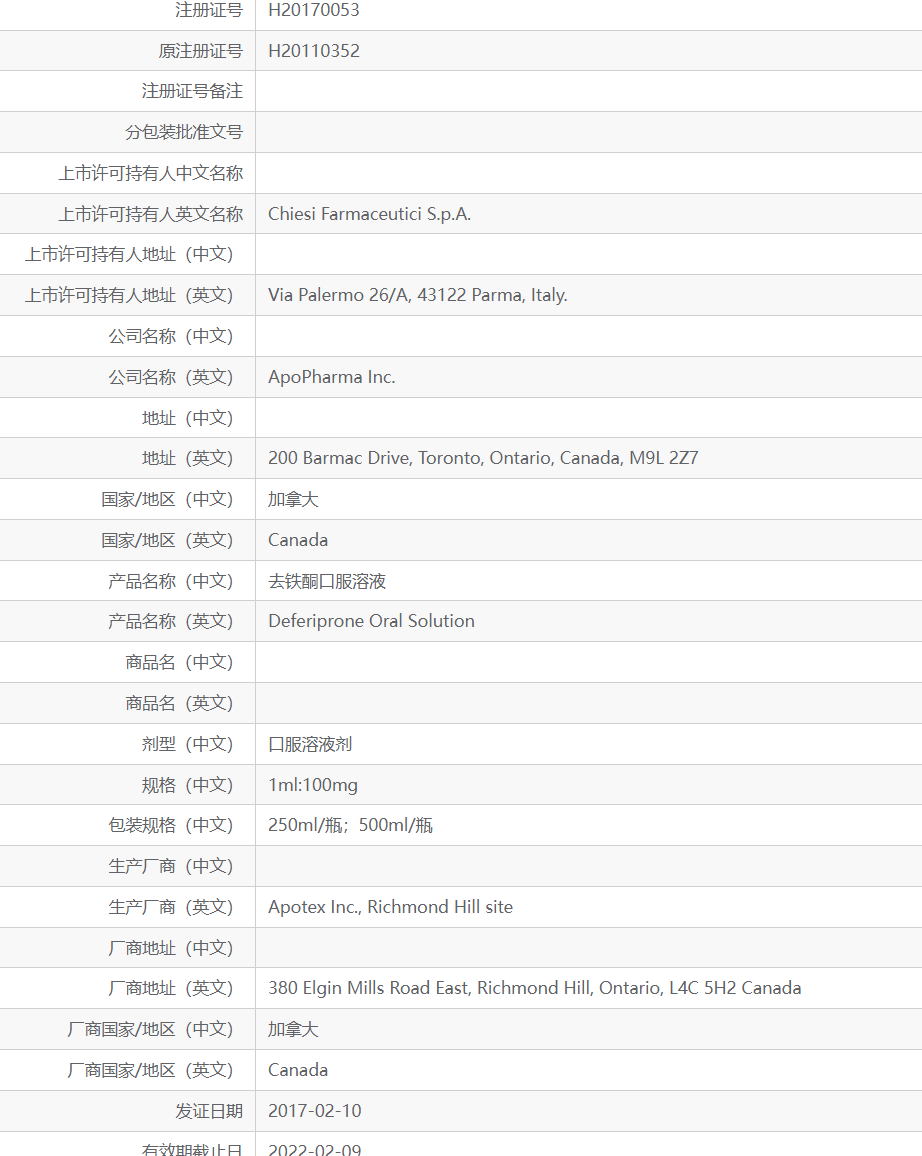
通过查询药品受理的数据库(药智),目前申报的情况两种情况都有。如下:
①去铁酮口服溶液在2017年获得进口再注册批准,当时是申请1个规格申报,分有2个包装规格。
②布洛芬混悬液,无论是强生的补充申请,还是新申请的仿制,都是按照2个规格申报。两个规格浓度相同,装量不同。比如强生,两个规格分别为30ml:0.6g和100ml:2g。
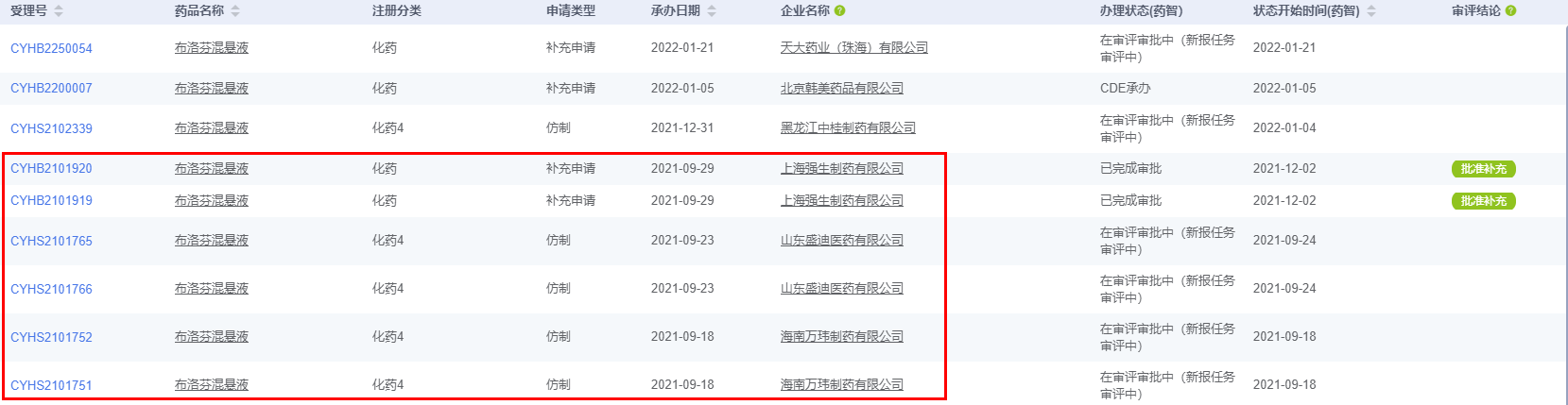
为什么都是混悬液不同的包装,却分别按照1个规格和2个规格申报?是不是我哪里理解有误。比如,报2个规格,是不是因为2个规格样品虽然浓度相同,但不是从同一药液分装而来(因为大小体积预期生产规模不一样),而报1个规格是因为不同包装来自同一批药液?不太了解混悬液(或者口服溶液)的实际生产过程,非常需要和欢迎有了解的老师给指点迷津。问这个问题是因为,如果按照2个规格申报,是否2个规格药品的稳定性研究是否也适用【建议】和【依据】里描述的方法?
建议:
1.根据ICH Q1A 新原料药和制剂稳定性试验 2.2.3 “批次的选择” 制剂的每一种规格和包装规格都应进行稳定性研究,除非应用了括号法和矩阵化设计。上述情况为不同包装规格的口服液体制剂 应逐规格进行稳定性试验:“应提供至少三个批次样品的稳定性资料,注册批次的处方和容器密封系统应与拟上市产品相同,其生产工艺应模拟上市产品的生 产工艺,其质量应与拟上市产品相同,符合相同的质量标准。如证明合理,其中两批必须至少在中试规模下生产,另一批可在较小规模下生产。可能的话,生产不同批次的制剂应采用不同批号的原料。
2.类似的指导原则见化学药物(原料药和制剂)稳定性研究技术指导原则(修订) 四、制剂的稳定性研究 “通常制剂的每一种规格和包装规格均应进行稳定性研究” ,具体如何选择:“注册申报时应提供至少3个注册批次制剂正式的稳定性研究资料。注册批次制剂的处方和包装应与拟上市产品相同,生产工艺应与拟上市产品相似,质量应与拟上市产品一致,并应符合相同的质量标准。如证明合理,新制剂3个注册批次其中2批必须至少在中试规模下生产,另1批可在较小规模下生产,但必须采用有代表性的关键生产步骤。仿制制剂3个注册批次均必须至少在中试规模下生产。在条件许可的情况下,生产不同批次的制剂应采用不同批次的原料药。”
依据:ICH Q1A 新原料药和制剂稳定性试验 2.2.3 “批次的选择” 及 《化学药物(原料药和制剂)稳定性研究技术指导原则(修订)》 “四、制剂的稳定性研究”
这{{threadTextType}}正{{isAdminText}}
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: