各位老师,
关于委托生产,
生物制品,新药,临床阶段,临床早期(Ib期),中美双报,临床样品生产场地变更,经过药品质量检测对比,工艺对比,药学具有可比性,是否可以不报补充申请?
由于中美双报,美国的直接上临床,中国的是否不报补充申请也可以直接上临床?(临床药品既在中国医院做试验,也在美国做临床试验)。
而且我们在中国的权益准备卖掉(BD给别的公司),也就是说以后也不打算在中国申报上市了,只在海外上市,这种情况,是否可以不在中国不报补充申请? 反正美国又不用报补充申请。
先上结论:生物制品临床试验期间的生产场地实质性变更,中美都需要向监管机构申报.。不过美国不叫补充申请,而叫信息补充(information amendment );只是从法理上说美国的CMC information amendment交上去后,场地变更后生产的样品就可以用在临床上(实践上许多公司会等30天,看FDA是否有异议)。
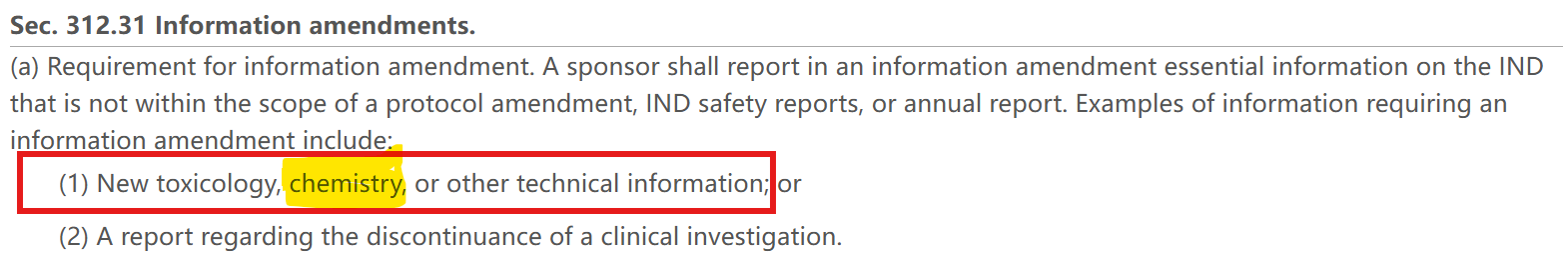
1. 美国: 21 CFR §312.31
生产场地的实质性变更行业内基本上都认为是触发CMC information amendment的。
2. 中国:《临床试验期间生物制品药学研究和变更技术指导原则(试行)》
虽然指导原则正文中说申办者评估认为不影响受试者安全的,可以直接实施,但在后面“可能增加受试者安全性风险”的事项中,“生产场地的实质性变更”赫然在列。通常实践上也都是交临床试验期间的药学补充申请的。

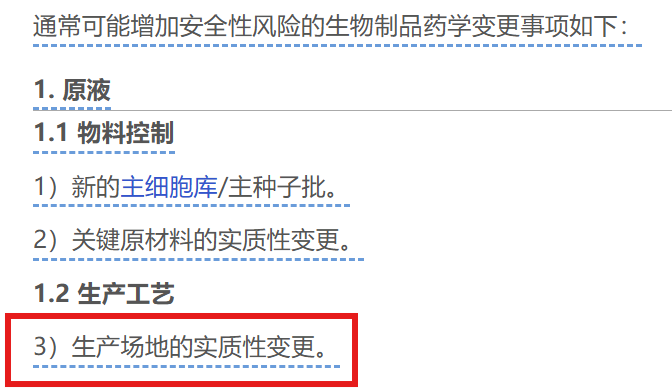
(提问者没提到,但为了说明临床试验期间的生产场地的实质性变更各监管机构的态意见基本一致,也补充上。)
生产场地变更也在最后那个substantial modifications 的表中,即需要对IMPD进行信息修订。

最后吐槽一下:啥叫中国权益卖掉,以后不打算在中国申报上市了?人家BD个寂寞么? 不能坑买你们中国权益的公司啊!
这{{threadTextType}}正{{isAdminText}}
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: