制药领域几乎众所周知,欧盟药监局(EMA)的现行版法规文件-GMP
Annex 1【1】(即欧盟GMP附录1,以下简称附录1)中要求对除菌级过滤器进行使用前、灭菌后完整性测试(PUPSIT,Pre-Use
and Post-Sterilization Integrity Test)。
本文内容:
1.
为什么要做除菌过滤器/工艺的使用前、灭菌后的完整性检测(PUPSIT)?
(Why) Regulatory Requirements
for Pre-Use and Post-Sterilization Integrity Test (PUPSIT) of Sterile
Filtration Process
2.
如何以良好实践经验为基础完成 PUPSIT?
(How)Implementation
of the Design and Use of PUPSIT based on GMP
3.
如何安全合理的替代执行PUPSIT?
(What) Strategies to Replace the PUPSIT
基于附录1里的相关要求,凡是需要接受欧盟药监部门监管的制药企业基本都要接受其规定并执行。在我国,NMPA在2018年颁布的《除菌过滤技术及应用指南》【3】当中对使用前的除菌过滤器完整性检测也有相关要求,如“6.3 完整性测试 除菌过滤器使用后,必须采用适当的方法立即对其完整性进行测试并记录。除菌过滤器使用前,应当进行风险评估来确定是否进行完整性测试,并确定在灭菌前还是灭菌后进行。……”。由此可见法规对实施相关设计及操作有明确的要求。
2.
如何以良好实践经验为基础完成 PUPSIT ?
以冗余除菌过滤设置*为例,为保证过滤工艺开始之前既能有效实施PUPSIT检测、同时又要尽可能降低检测过程对过滤系统引入微生物污染的风险,因此无论是以不锈钢材质组件还是一次性使用技术(SUT,Single Use Technology)搭建的过滤系统,都无法避免在管路及相关组件的设计上的复杂性。为了帮助业界解决上述设计复杂性的问题,如下管路设计图所示,我们结合相应的工程要求及实践经验而设计出了可以便捷执行PUPSIT的SUT冗余除菌过滤系统。无论在检测前对过滤系统进行液体润湿、排放润湿液,还是在实施PUPSIT检测过程时对每一级过滤器单独进行检测完整性,该系统都能有效且简便的实现相关操作。
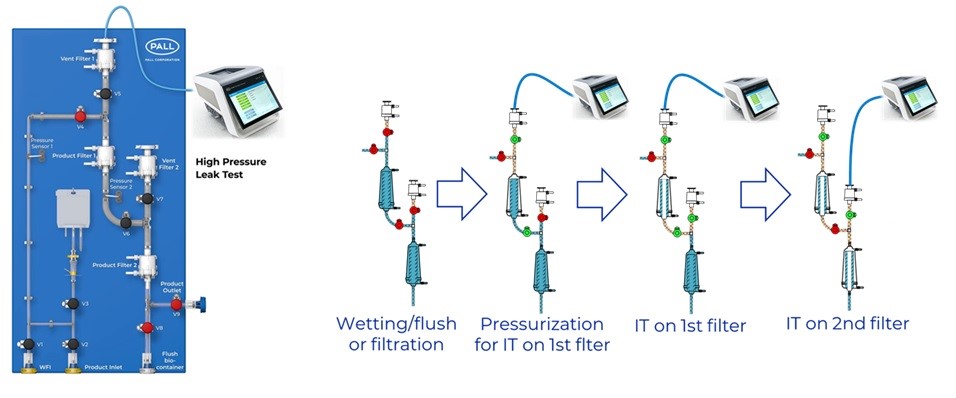
*
“通常通过两个或以上相同或递减孔径的过滤方式,统称为序列过滤。序列过滤系统中,如果在最终除菌过滤器前增加一个除菌级过滤器,并且确保两个过滤器之间无菌,以及控制过滤前介质的微生物污染水平一般小于等于10cfu/100ml,这种情况下称为冗余过滤系统。”【3】

除了上述过滤系统展示了使用膜面积较小的除菌过滤器所搭建的系统,如此图所示,我们另外还为具有相对较大膜面积的过滤工艺开发了较大规模的PUPSIT过滤系统。
3. 如何安全合理的替代执行PUPSIT
PUPSIT之所以被EMA高度重视,前文已经提到了出于防范因灭菌而引入损坏风险的原因,这里我们关注行业内另外一个热议的问题:即“Flawed Filter Masking”【5】,这个概念大致是指经灭菌处理后的除菌过滤器可能存在微小的缺陷,导致虽然其可以顺利通过完整性测试、但却有微生物穿透的风险。
为了以实际数据来揭示“Flawed Filter Masking”发生的具体情况,2017年PDA与BPOG就PUPSIT话题联合成立了 SFQRM专家组(Sterile Filtration
Quality Risk Management Consortium,除菌过滤质量风险管理联盟)【2】,组内部分专家后续在相关杂志上发表过相应的研究结果。专家们针对“Flawed
Filter Masking”的问题首先提出了两个假设条件,即:
1. 滤膜上存在有缺陷的膜孔(大到可以穿透细菌、小到过滤过程可将其堵塞);
2. 料液可以堵塞有缺陷的膜孔、并能使其顺利通过完整性检测。
经研究得知【5】:在过滤器堵塞程度大于81%的测试中的确会有小概率的“Flawed Filter
Masking”现象出现。换言之,在相对较高的污染物堵塞的状态下,过滤器的确是存在有缺陷的膜孔被掩盖的风险,导致完整性检测无法识别风险。基于研究,专家们总结出一些有指导意义的信息及应对策略:
Ø 可滤性实验(小试研究)很重要,可以帮助降低“Flawed Filter Masking”问题发生的概率,如添加合适的预过滤工艺以降低待过滤流体中的污染物含量,从而降低 除菌过滤工艺中的高污堵的概率;
Ø 制药企业应基于风险评估来决定PUPSIT实施与否,比直接强制执行将会是更好的选择。
基于上述应对方法,在附录 1的修订过程中,以充分的风险评估的方式来替代PUPSIT则成为了一种可能,附录 1 的修订版中也可见相关说明。我们的专家团队结合多年来的实践经验,归纳总结出以下几个维度信息(包括但不限于)以供行业进行评估时参考使用:
过滤器的生产
过滤器的运输、储存、拆包及安装
对过滤器的灭菌
过滤器的设置
待过滤的产品
截止此文发布时,欧盟GMP附录 1还处于草稿发布阶段,当前的草案对风险评估的替代方式显示出一定的接受度和灵活性,但同样有业界声音表示:预计在2023年9月将以定稿发布的附录 1中,虽然提及有可“豁免”的情况,却仍有可能在很大程度上保留倡导执行PUPSIT的原则。
参考文献
【1】
EUROPEAN COMMISSION, EudraLex, Volume 4 - EU
Guidelines to Good Manufacturing Practice, Annex 1 - Manufacture of Sterile
Medicinal Products(November 2008)
【2】
Points to Consider for Risks Associated with
Sterilizing Grade Filters and Sterilizing Filtration, PDA-BioPhorum 2020,
ISBN:978-1-945584-17-6
【3】
国家药品监督管理局2018年第85号通告附件1- 《除菌过滤技术及应用指南》
【4】
《中国药典》(2020版)三部-通则和指导原则-其它-1421
灭菌法-过滤除菌法
【5】
Test Process and Results of Potential Masking of
Sterilizing Grade Filters, PDA Journal
of Pharmaceutical Science and Technology 2020,74(5):509-523.DOI:10.5731/pdajpst.2019.011189.
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: